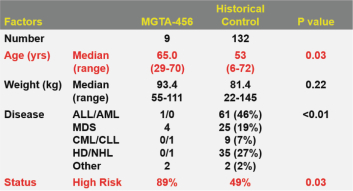
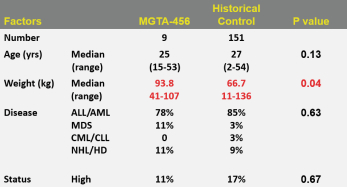
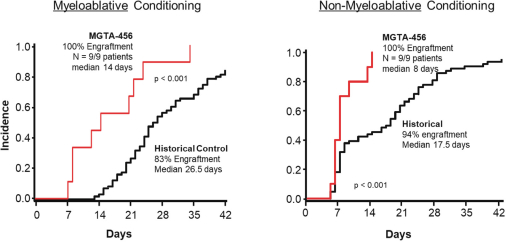
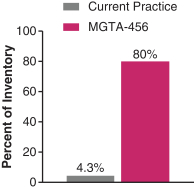
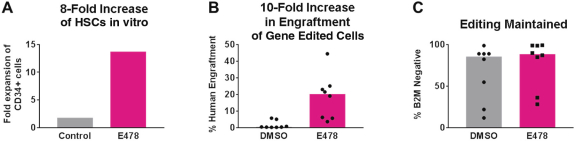
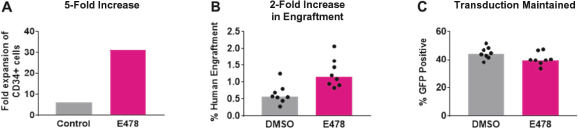
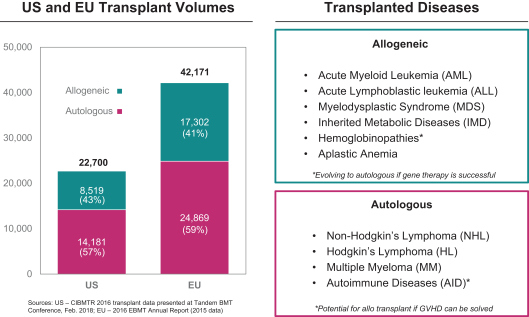
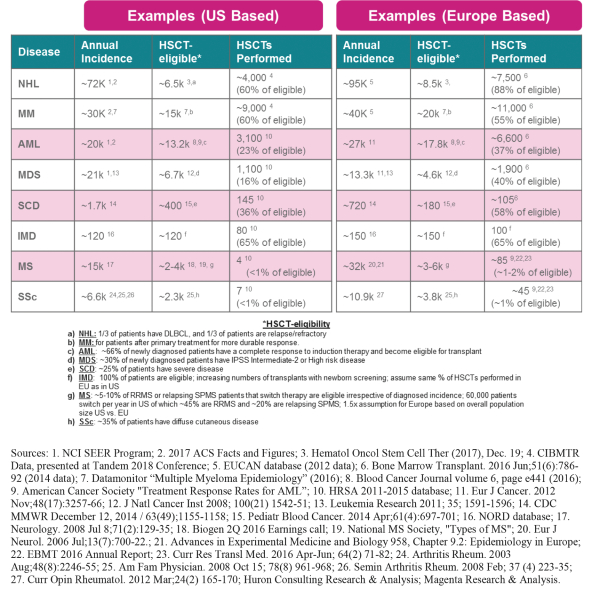
Even if we, or any collaborators we may have, obtain marketing approvals for any product candidates we
develop, the terms of approvals and ongoing regulation of our products could require the substantial expenditure of resources and may limit how we, or they, manufacture and market our products, which could materially impair our ability to generate
revenue.
Any product candidate for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data,
labeling, advertising, and promotional activities for such medicine, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing
information and reports, registration and listing requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, and requirements regarding the distribution of samples to
physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the medicine may be marketed or to the conditions of approval, or contain
requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the medicine.
Accordingly, assuming we, or any
collaborators we may have, receive marketing approval for one or more product candidates we develop, we, and such collaborators, and our and their contract manufacturers will continue to expend time, money, and effort in all areas of regulatory
compliance, including manufacturing, production, product surveillance, and quality control. If we and such collaborators are not able to comply with post-approval regulatory requirements, we and such collaborators could have the marketing approvals
for our products withdrawn by regulatory authorities and our, or such collaborators’, ability to market any future products could be limited, which could adversely affect our ability to achieve or sustain profitability. Further, the cost of
compliance with post-approval regulations may have a negative effect on our business, operating results, financial condition, and prospects.
Risks Related to Intellectual Property
We are highly dependent on intellectual property licensed from third parties and termination of any of these licenses could result in the loss of
significant rights, which would harm our business.
In April 2017, we entered into a license agreement with Novartis pursuant to which we were
granted a worldwide license to certain intellectual property rights owned or controlled by Novartis, including patents, patent applications, proprietary information, know-how and other intellectual
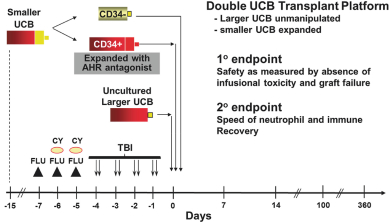
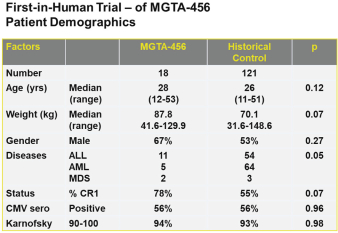
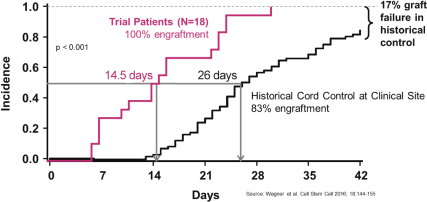
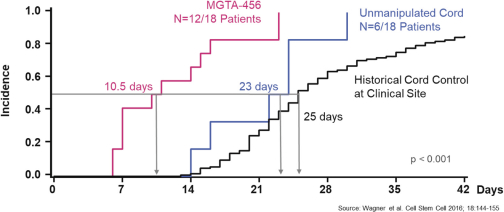
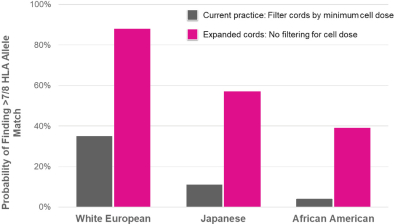
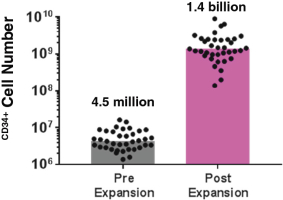
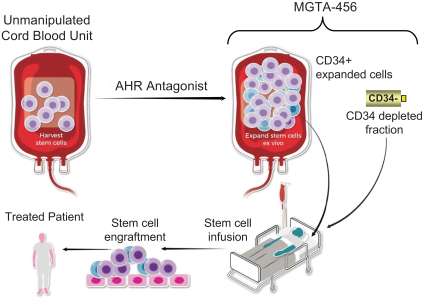
property, to develop, commercialize and sell one or more therapeutic products comprising MGTA-456 in the field of non-gene-edited/-modified HSCs. In addition, in
November 2016, we entered into a license agreement with Harvard University, or Harvard, pursuant to which we were granted a worldwide license to research, develop and commercialize one or more therapeutic products under certain conditioning- and
mobilization-related patents and patent applications owned or controlled by Harvard. Furthermore, in March 2018, we entered into a research, development option and license agreement with Heidelberg Pharma Research GmbH, or Heidelberg Pharma,
pursuant to which we intend to combine our proprietary antibodies and Heidelberg Pharma’s amanitin conjugates platform. We are dependent on the patents, know-how and proprietary technology, licensed from
Novartis and Harvard. Furthermore, if we commercialize any products utilizing Heidelberg Pharma’s amanitin conjugates platform, we will be dependent on the intellectual property rights we license from Heidelberg Pharma. Any termination of these
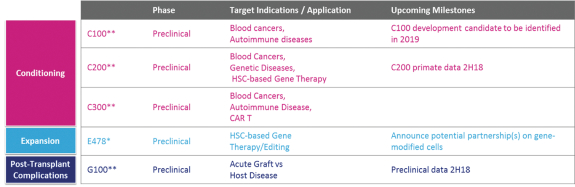
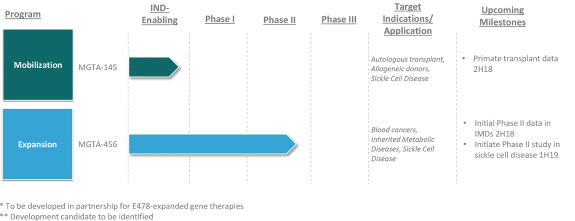
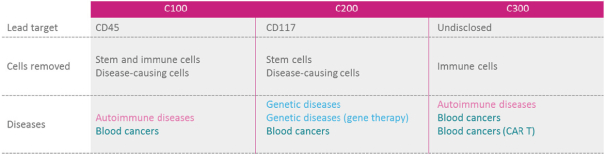
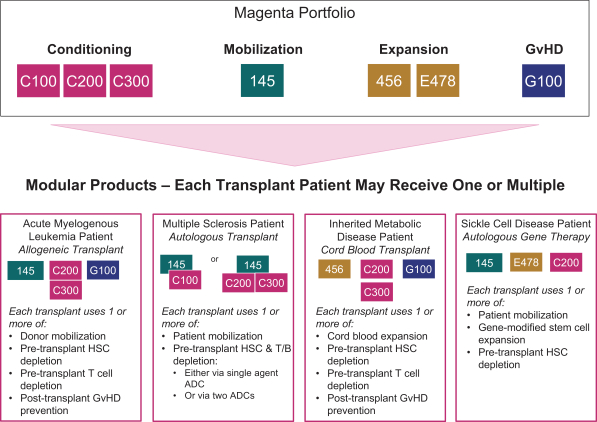
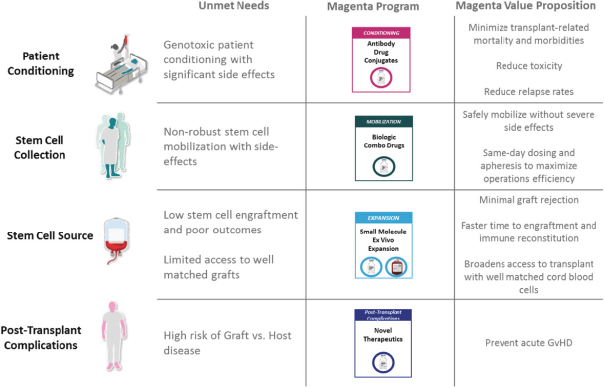
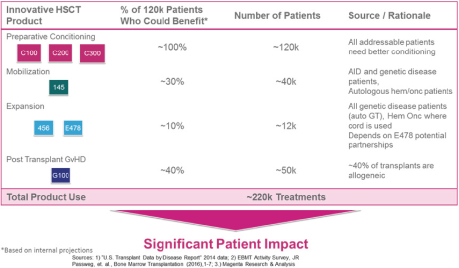
licenses, or a finding that such intellectual property lacks legal effect, could result in the loss of significant rights and could harm our ability to commercialize MGTA-456, C100, C200, C300, G100, MGTA-145, and other product candidates.
Certain of our license agreements, including our agreements with Novartis,
Harvard and Heidelberg Pharma, require us to use diligent efforts or meet development thresholds to maintain the license, including establishing a set timeline for developing and commercializing products. If we fail to comply with the obligations
under our license agreements, including payment terms and diligence terms, our licensors may have the right to terminate our agreements, in which event we may not be able to develop, manufacture, market or sell the products covered by our agreements
or may face other penalties under our agreements. Such an occurrence could materially
40